Data Generation
The triplicated and quadruplicated human gene families residing on four paralogons ( Hsa 2/7/12/17, Hsa 4/5/8/10, Hsa 1/2/8/20 and Hsa 1/6/9/19) were identified by chromosomal scanning using Ensembl and UCSC genome browsers (Hubbard al., 2002). The closest putative orthologous sequences of the human proteins in other species were obtained using BLASTP in the Ensembl genome browser (Clamp et al., 2003; Hubbard et al.,2002) and enrichment of data was conducted by using protein database available at the National Center for Biotechnology Information (Hughes et al., 2001a; Johnson et al., 2008) and the Joint Genome Institute (http://www.jgi.doe.gov/).
Amino acid sequences were aligned using a multiple sequence alignment tool CLUSTAL-W with default parameter (Thompson et al., 1994). The phylogenetic analysis was performed by reconstructing neighbor joining (NJ) (Russo et al., 1996; Saitou and Nei, 1987) and Maximum Likelihood (Whelan and Goldman, 2001) trees using MEGA version 5 (Kumar et al., 2008). The co-duplicated groups were detected through topology comparison approach and reflect the branching pattern of phylogenetic trees. The tree topologies of gene families were compared to validate consistency in duplication events (Zhang and Nei, 1996). The age estimates of duplication events of multigene families residing on each paralogon were evaluated using relative dating approach.
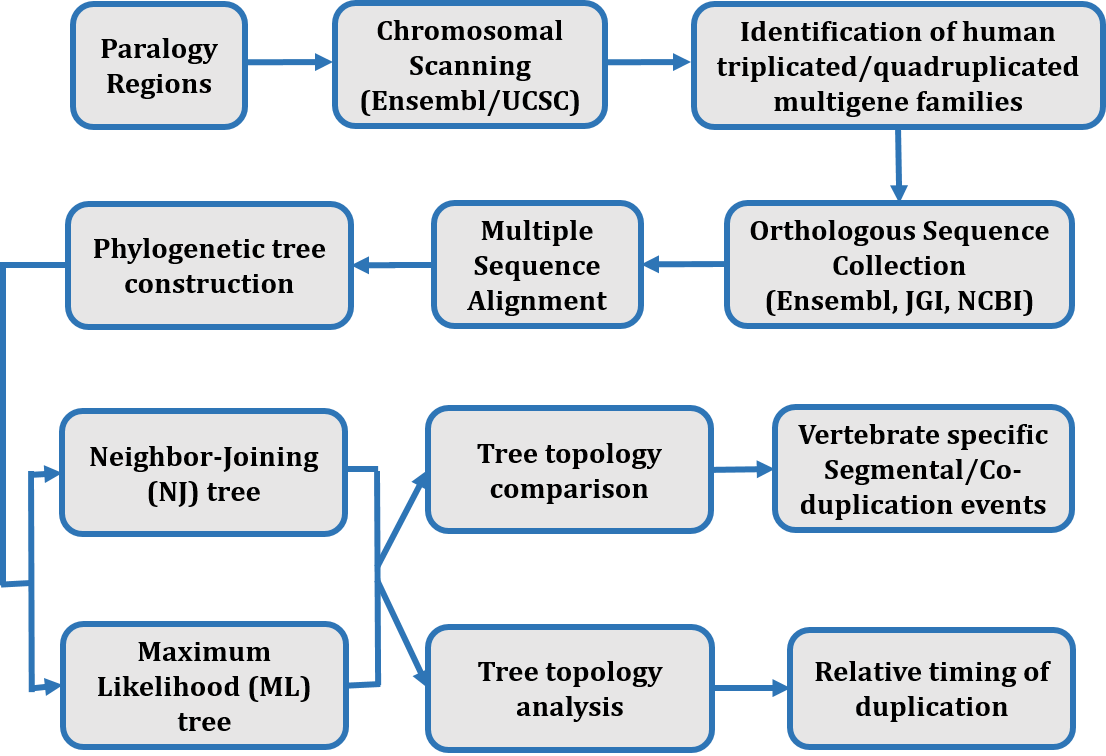
Figure 1: Flow of Data generation
Useful references:
Clamp, M., Andrews, D., Barker, D., Bevan, P., Cameron, G., Chen, Y., Clark, L., Cox, T.,Cuff, J., Curwen, V., Down, T., Durbin, R., Eyras, E., Gilbert, J., Hammond, M., Hubbard, T., Kasprzyk, A., Keefe, D., Lehvaslaiho, H., Iyer, V., Melsopp, C., Mongin, E., Pettett, R., Potter, S., Rust, A., Schmidt, E., Searle, S., Slater, G., Smith, Birney, E., (2003) Ensem Felsenstein J. 1985. Confidence-Limits on Phylogenies - an Approach Using the Bootstrap. Evolution, 39, 783-791.
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, Gilbert J, Hammond M, Huminiecki L, Kasprzyk A, Lehvaslaiho H, Lijnzaad P, Melsopp C, Mongin E, Pettett R, Pocock M, Potter S, Rust A, Schmidt E, Searle S, Slater G, Smith J, Spooner W, Stabenau A, Stalker J, Stupka E, Ureta-Vidal A, Vastrik I, Clamp M. (2002) The Ensembl genome database project. Nucleic Acids Research, 30, 38-41.
Hughes, A.L., da Silva, J., Friedman, R., (2001) Ancient genome duplications did not structure the human Hox-bearing chromosomes. Genome Res, 11, 771–780.
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. (2008) NCBI BLAST: a better web interface. Nucleic Acids Research, 36, W5-9.
Kumar S, Nei M, Dudley J, Tamura K. (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform, 9, 299-306.
Russo, C.A., Takezaki, N., Nei, M., (1996) Efficiencies of different genes and different tree-building methods in recovering a known vertebrate phylogeny. Mol. Biol. Evol, 13, 525–536.
Saitou, N., Nei, M., (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol, 4, 406–425.
Thompson JD, Higgins DG, Gibson TJ. (1994) Clustal-W - Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Research, 22, 4673-4680.
Whelan, S., Goldman, N., (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach.
Zhang JZ, Nei M. (1996) Evolution of antennapedia-class homeobox genes. Genetics, 142, 295-303.